
Sales & marketing people are, in general, not very much interested in regulatory stuff. However, if you do not want your sales in the UK and Europe to be disrupted, it is important to gain knowledge of the changes that will take place very soon.
There are already numerous articles about the changes, guidelines, and protocols of the MDR regulations, so we won’t bore you with them. A few websites you can find more information on are listed below the sections.
In this blog, we cover how EUMDR and UK medical device regulations affect your European importers and distributors.
Selling Medical Devices into Europe Via Distributors/Importers: Understanding EUMDR
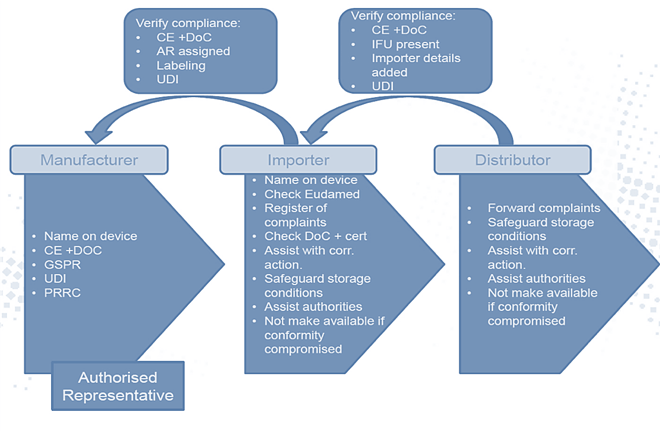
For medical device manufacturers selling into Europe under the CE mark, changes will take place in the liabilities and responsibilities of the manufacturer and the economic operators (European importers and distributors, EO). (Figure 1)
The goal of this change is to make each entity responsible and serve as a control/safety check for the products introduced in the European market and organizations involved are compliant with the MDR.
Your distributor may be your importer, you may be the importer, or you may choose to have a separate importer and distributor. There are multiple ways to organize the pros and cons depending on your agreements, the size of your European presence, and resources.
It is the manufacturer’s responsibility to map out the EO chain to define the operating identities in Europe. Failing to do so will put the business at significant risk of compliance and non-conformities.
How medical device regulations affect European importers
Importers, like distributors, had no regulatory obligations. Under the new medical device regulations, it has changed. Now, they are responsible for conducting specific checks necessary to ensure the products they represent are compliant by checking CE marks, declarations of conformity, and registration in EUDAMED and by identifying the manufacturer and authorized representative.
Furthermore, they have to ensure systems and processes are in place that comply with the new requirement to:
- Keep a register of complaints,
- Non-conforming devices,
- Recalls and withdrawals to enable investigation by the competent
authorities if required, - Forward complaint information to manufacturers,
- Notification system for non-compliant devices to the manufacturer,
the competent authority, and an authorized representative, - Ensure storage and maintenance conditions are respected and check
that decisions are labeled and always match their instructions for use (IFSs),
and UDI labels are present where needed.
Importers must ensure devices are labeled with information allowing for importer identification:
- The importer’s name,
- Importers' trade name or trademark,
- Registered place of business,
- Address for contact,
- No obscuration of the manufacturer's label.

This transition and verifying whether the manufacturer located outside the EU has the technical, scientific, and financial capacity to continue manufacturing an EU-compliant device.
Medical device regulations' impact on European distributor obligations
Distributors did not face any regulatory obligations in the past either. In the new medical device regulations, they are jointly and severally liable to:
- Ensure storage or transport conditions comply with those set out by the
manufacturer, - Verify Instruction For Use (IFU) is included with each device, and the CE
mark, declaration of conformity, and any UDI’s are present, - Taking corrective action and notification to the Competent Authority,
manufacturer, and authorized representative when a device presents a
serious risk or is falsified, - Authority with information, samples, and access to the device,
- Forwarding complaints and keeping a register of complaints, non-conforming
device recalls, and withdrawals.
The main additional role of the distributor is to include verification processes that verify the CE mark, EU declaration of conformity, IFUs, compliance of importer, and the right UDI assigned to the device by the manufacturer.
.jpeg?width=1920&height=1080&name=Template%20%20%231%20(1).jpeg)
The UKCA Mark
The UKCA (UK conformity assessed) mark is the newly introduced UK product marking that will be used for certain goods, including medical devices, being placed on the Great Britain market.
Manufacturers of devices that will be placed on the Great Britain market will be able to use this mark starting from the end of the transition period, i.e. 1st January 2021. The MHRA will continue to recognize the CE mark until the 1st of July 2023 for devices placed on the Great Britain market that have been CE marked under and fully conform with any of the current Directives or incoming Regulations of the EU for Medical Devices (active implantable medical devices) and IVD devices.
The UKCA mark will not, however, be recognized in the EU, EEA, and NIR markets, and as such, it will not be interchangeable with the CE mark outside of Great Britain. To this effect, devices that will be placed on those markets will continue to need a CE mark for this purpose.
From the 1st of July 2023, the UKCA will be the only marking recognized in Great Britain, and as such, all manufacturers will need to meet the relevant requirements and place the UKCA mark on their devices.

Labeling requirement changes under UK medical device regulations
Devices placed on the Great Britain market after the 1st of January 2021 may either carry the CE mark or the UKCA mark, depending on which legislation has been used as the basis for the conformity assessment and certification of the device. The number of the NB or UK-approved body will also have to appear on the label, where applicable.
Devices that will carry a valid CE mark at the end of the transition period will not be required to be re-labeled with the UKCA until the 1st of July 2023. During this time, they may be placed on the Great Britain market with the CE mark on their label. Devices that carry a dual mark on their label can continue to be sold on the Great Britain market after the 1st of July 2023 by virtue of carrying the UKCA mark, irrespective of the CE mark also appearing on the label.
The presented proposals will take effect through legislative changes introduced later this year and are, therefore, still subject to parliamentary approval.
UK MDR Recommendations and Challenges
These are all new processes for your distributors and importers. It is important to educate them on these changes, as many are still not aware.
Discuss and requalify their capabilities and resources to determine whether they will and can comply. If, in an audit, it comes to light they are not in compliance, you, as a manufacturer, will be affected due to not being able to use this channel and experience disruptions in sales.
Build in annual performance reviews that evaluate these processes and involve the regulatory responsible person as well. Keep a record of these meetings and make sure the documents required for distributors and importers are easily accessible.
Above all, it is important there is a contract in place that is compliant with the UK medical device regulations and formalizes all agreements between the parties. More information can also be found on www.mdlaw.eu.
Feel free to reach out for our Sales Outsourcing solutions in the European medical device sector.
Sources:

Related articles
-

MDR vs. FDA 510(k): Where U.S. Playbooks Break in Europe (and How to Fix Them)
Last updated: 2 March 2026Learn the key differences between FDA 510(k) and EU MDR, including classification changes, clinical...
Read more -

Expanding Non-Invasive Monitoring Tech into Europe: A Path for North American Innovators
Last updated: 26 August 2025Europe’s healthcare systems need scalable, non-invasive monitoring tools. Discover key...
Read more -

Navigating the European Market: Challenges and Opportunities for American Medical Companies
Last updated: 23 July 2025Europe’s medical device market offers huge growth potential for American MedTech companies, but...
Read more